
Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
Huntingtonova choroba
Lékařský expert článku

Naposledy posuzováno: 05.07.2025
Huntingtonova choroba je autozomálně dominantní neurodegenerativní onemocnění charakterizované progresivním kognitivním poklesem, mimovolními pohyby a zhoršenou motorickou koordinací, které začíná ve středním věku. Diagnóza se potvrzuje genetickým testováním. Léčba je primárně symptomatická. Genetické testování může být doporučeno u pokrevních příbuzných. George Huntington poprvé popsal toto onemocnění v roce 1872 poté, co studoval rodinný případ u obyvatel Long Islandu.
Prevalence Huntingtonovy choroby je přibližně 10 případů na 100 000 obyvatel a vzhledem k jejímu pozdnímu nástupu má přibližně 30 lidí ze 100 000 50% riziko, že se u nich během života rozvine. Ačkoli se onemocnění nejčastěji objevuje mezi 35. a 40. rokem života, věkové rozpětí nástupu je poměrně široké, přičemž nejranější nástup je ve 3. roce života a nejpozději v 90. roce života. Ačkoli se původně předpokládalo, že onemocnění má 100% penetranci, nyní se předpokládá, že tomu tak vždy není. U jedinců, kteří zdědili gen pro toto onemocnění po svém otci, se onemocnění projeví v průměru o 3 roky dříve než u těch, kteří zdědili patologický gen po matce. U přibližně 80 % pacientů, kteří zdědili patologický gen po svém otci, se onemocnění projeví před 20. rokem věku. Fenomén dřívějšího projevu genetické vady u potomků se nazývá anticipace.
 [ 1 ]
[ 1 ]
Co způsobuje Huntingtonovu chorobu?
Huntingtonova choroba nemá genderovou preferenci. Projevuje se atrofií nucleus caudatus, kde dochází k degeneraci malých neuronů a klesá hladina neurotransmiterů - kyseliny gama-aminomáselné (GABA) a substance P.
Za rozvoj Huntingtonovy choroby je zodpovědný mutantní gen se zvýšeným počtem („expanzí“) sekvencí DNA CAG (cystein-alanin-glycin) kódujících aminokyselinu glutamin. Produkt tohoto genu, velký protein huntingtin, obsahuje nadměrné množství polyglutaminových zbytků, což vede k onemocnění neznámým mechanismem. Čím více CAG repetic, tím dříve onemocnění debutuje a tím závažnější je jeho průběh. Z generace na generaci se počet repetic může zvyšovat, což časem vede ke zhoršení rodinného fenotypu.
Navzdory značnému zájmu o genetické a biochemické změny u Parkinsonovy choroby bylo hledání genu pro toto onemocnění neúspěšné až do konce 70. let 20. století. V té době Nancy Wexlerová a Allan Tobin uspořádali workshop sponzorovaný Nadací pro dědičné choroby, aby prodiskutovali strategii pro nalezení genu pro Huntingtonovu chorobu. David Housman, David Botstein a Ray White, kteří se setkání zúčastnili, navrhli, že by nedávno vyvinuté techniky rekombinantní DNA mohly pomoci dosáhnout tohoto cíle. Klíčovým úkolem projektu bylo najít velkou rodinu s mnoha generacemi Huntingtonovy choroby za účelem získání vzorků DNA. V roce 1979 byl zahájen společný projekt vědců z Venezuely a Spojených států, jehož cílem bylo vyšetřit velkou rodinu s Huntingtonovou chorobou žijící na břehu jezera Maracheibo (Venezuela). V roce 1983 byl gen Huntingtonovy choroby lokalizován na konci krátkého raménka chromozomu 4 (Gusella a kol., 1983) a o deset let později bylo zjištěno, že mutace tohoto genu spočívá ve zvýšení počtu opakování trinukleotidu cytosin-adenin-guanin (CAG) (Huntington's Disease Collaborative Research Group, 1993). Metodologie vyvinutá touto vědeckou skupinou je v současnosti považována za standardní pro poziční klonování nových genů.
Zatímco gen divokého typu má úsek 10-28 CAG repetic, mutantní forma genu, který způsobuje Huntingtonovu chorobu, má zvýšený úsek z 39 na více než 100 CAG repetic. Objev expanze trinukleotidových repetic pomohl vysvětlit mnoho klinických rysů onemocnění. Zejména byla zjištěna inverzní korelace mezi věkem nástupu a délkou oblasti s opakovanými trinukleotidy. Předpokládanou otcovskou dědičnost lze vysvětlit skutečností, že u mužů se během spermatogeneze často vyskytuje zvýšení počtu repetic. Analýza nových mutací ukázala, že k nim obvykle dochází, když jeden z rodičů, obvykle otec, měl počet CAG repetic vyšší než 28; v tomto případě se počet těchto repetic v další generaci zvýšil. Nyní bylo zjištěno, že pokud počet repetic není vyšší než 28, je stabilně přenášen z generace na generaci. Pokud je počet opakování od 29 do 35, pak se příznaky Huntingtonovy choroby neprojevují, ale při přenosu na potomky se délka této oblasti může zvětšit. Pokud je počet opakování od 36 do 39, pak se v některých případech (ale ne vždy) může onemocnění projevit klinicky (neúplná penetrance) a při přenosu na potomky je možné zvýšení počtu trinukleotidových opakování. Pokud počet opakování přesáhne 40, pak se onemocnění vyskytuje téměř ve všech případech a při přenosu na potomky je možné další rozšíření opakování. Důvody zvýšení počtu opakování zůstávají neznámé.
Patomorfologie Huntingtonovy choroby
Huntingtonova choroba je charakterizována ztrátou neuronů převážně v nucleus caudatus a putamen a do určité míry i v kortexu a dalších mozkových strukturách. Celková hmotnost mozku je u Huntingtonovy choroby snížena nejen snížením počtu neuronů, ale také ztrátou bílé hmoty. V mozkové kůře jsou nejvíce postiženy buňky ve vrstvách V a VI. Závažnost mikro- a makroskopických degenerativních změn (upraveno na věk v době úmrtí) koreluje s počtem CAG repetic. Detailní patologická analýza změn u několika stovek případů Huntingtonovy choroby ukázala, že degenerace striata začíná v dorsomediální části nucleus caudatus a dorsolaterální části putamen a poté se šíří ventrálně. Různé skupiny neuronů v nucleus caudatus a putamen jsou postiženy v různé míře. Interneurony ve striatu zůstávají relativně intaktní, ale některé projekční neurony jsou selektivně postiženy. U juvenilní formy Huntingtonovy choroby jsou patomorfologické změny ve striatu výraznější a rozšířenější, postihují mozkovou kůru, mozeček, thalamus a globus pallidus.
Neurochemické změny u Huntingtonovy choroby
GABA. Neurochemické studie mozku u pacientů s Huntingtonovou chorobou odhalily významný pokles koncentrace GABA ve striatu. Následné studie potvrdily, že Huntingtonova choroba je spojena se snížením počtu GABAergních neuronů a ukázaly, že koncentrace GABA jsou sníženy nejen ve striatu, ale také v jeho projekčních zónách - vnějších a vnitřních segmentech globus pallidus a substantia nigra. V mozku u Huntingtonovy choroby byly také detekovány změny v receptorech GABA pomocí studií vazby receptorů a in situ hybridizace mRNA. Počet receptorů GABA byl mírně snížen v nucleus caudatus a putamen, ale zvýšen v retikulární části substantia nigra a vnějším segmentu globus pallidus, což je pravděpodobně způsobeno denervační přecitlivělostí.
Acetylcholin. Acetylcholin je využíván jako neurotransmiter velkými neostnitými interneurony ve striatu. Rané postmortální studie u pacientů s Huntingtonovou chorobou prokázaly sníženou aktivitu cholin acetyltransferázy (ChAT) ve striatu, což naznačuje ztrátu cholinergních neuronů. Ve srovnání s významným snížením GABAergních neuronů jsou však cholinergní interneurony relativně ušetřeny. Proto je hustota acetylcholinesterázově pozitivních neuronů a aktivita ChAT ve striatu ve skutečnosti relativně vyšší ve srovnání s kontrolní skupinou stejného věku.
Látka P. Látka P je obsažena v mnoha středně ostnatých neuronech striata, které převážně vyčnívají do vnitřního segmentu globus pallidus a substantia nigra a obvykle také obsahují dynorfin a GABA. Hladiny látky P ve striatu a pars reticularis substantia nigra jsou u Huntingtonovy choroby sníženy. V terminálním stádiu onemocnění imunohistochemické studie odhalily významné snížení počtu neuronů obsahujících látku P. V dřívějších stádiích jsou neurony obsahující látku P a vyčnívající do vnitřního segmentu globus pallidus relativně ušetřeny ve srovnání s neurony vyčnívajícími do pars reticularis substantia nigra.
Opioidní peptidy. Enkefalin je obsažen ve středních ostnatých výběžcích GABAergních neuronů nepřímé dráhy, které vyčnívají do vnějšího segmentu globus pallidus a nesou D2 receptory. Imunohistochemické studie ukázaly, že neurony obsahující enkefalin vyčnívající do vnějšího segmentu globus pallidus se u Huntingtonovy choroby ztrácejí brzy. Tyto buňky zřejmě odumírají dříve než buňky obsahující substanci P vyčnívající do vnitřního segmentu globus pallidus.
Katecholaminy. Neurony obsahující biogenní aminy (dopamin, serotonin) a promítané do striata se nacházejí v kompaktní části substantia nigra, ventrálním tegmentu a raphe nuclei. Zatímco noradrenergní projekce do lidského striata jsou minimální, hladiny serotoninu a dopaminu (na gram tkáně) ve striatu jsou zvýšené, což naznačuje zachování těchto aferentních projekcí navzdory výrazné ztrátě vlastních neuronů striata. Dopaminergní neurony substantia nigra zůstávají intaktní jak u klasické, tak u juvenilní formy Huntingtonovy choroby.
Somatostatin/neuropeptid Y a syntetáza oxidu dusnatého. Měření hladin somatostatinu a neuropeptidu Y ve striatu u Huntingtonovy choroby odhalilo 4–5násobné zvýšení ve srovnání s normálními tkáněmi. Imunohistochemické studie prokázaly absolutní zachování striatálních interneuronů obsahujících neuropeptid Y, somatostatin a syntetázu oxidu dusnatého. Tyto neurony jsou tedy rezistentní vůči patologickému procesu.
Excitační aminokyseliny. Bylo navrženo, že selektivní buněčná smrt u Huntingtonovy choroby je způsobena neurotoxickým účinkem vyvolaným glutamátem. Hladiny glutamátu a kyseliny chinolinové (endogenní neurotoxin, který je vedlejším produktem metabolismu serotoninu a agonista glutamátových receptorů) ve striatu u Huntingtonovy choroby jsou mírně změněny, ale nedávná studie s využitím MR spektroskopie odhalila zvýšení hladin glutamátu in vivo. Hladina gliového enzymu zodpovědného za syntézu kyseliny chinolinové ve striatu u Huntingtonovy choroby je přibližně 5krát zvýšena ve srovnání s normálem, zatímco aktivita enzymu, který zajišťuje degradaci kyseliny chinolinové, je u Huntingtonovy choroby zvýšena pouze o 20–50 %. Syntéza kyseliny chinolinové tedy může být u Huntingtonovy choroby zvýšena.
Studie receptorů excitačních aminokyselin (EAA) u Huntingtonovy choroby odhalily významné snížení počtu NMDA, AMPA, kainátových a metabotropních glutamátových receptorů ve striatu, stejně jako AMPA a kainátových receptorů v mozkové kůře. V pozdním stádiu Huntingtonovy choroby NMDA receptory prakticky chyběly, zatímco v preklinických a raných stádiích byl zaznamenán významný pokles počtu těchto receptorů.
Selektivní citlivost. U Huntingtonovy choroby dochází selektivně ke ztrátě určitých typů striatálních buněk. Neurony středního trnu, které vyčnívají do zevního segmentu globus pallidus a obsahují GABA a enkefalin, odumírají velmi brzy v průběhu onemocnění, stejně jako neurony obsahující GABA a substanci P a vyčnívající do retikulární části substantia nigra. Ztráta neuronů obsahujících GABA a enkefalin a vyčnívajících do zevního segmentu globus pallidus deinhibuje tuto strukturu, což následně vede k aktivní inhibici subtalamického jádra. Snížená aktivita subtalamického jádra může zřejmě vysvětlit choreiformní pohyby, ke kterým dochází u Huntingtonovy choroby. Již dlouho je známo, že fokální léze subtalamického jádra mohou způsobit choreu. Ztráta neuronů GABA a substance P vyčnívajících do substantia nigra pars reticularis je pravděpodobně zodpovědná za okulomotorické poruchy pozorované u Huntingtonovy choroby. Tato dráha normálně inhibuje neurony substantia nigra pars reticularis vyčnívající do superior colliculus, které následně regulují sakády. U juvenilní Huntingtonovy choroby jsou výše uvedené dráhy postiženy závažněji a navíc se brzy ztrácejí striatální projekce do vnitřního segmentu globus pallidus.
Protein huntingtin, kódovaný genem, jehož mutace způsobuje Huntingtonovu chorobu, se nachází v různých strukturách mozku a dalších tkáních. Huntingtin se normálně nachází převážně v cytoplazmě neuronů. Protein se nachází ve většině neuronů v mozku, ale nedávné údaje ukazují, že jeho obsah je vyšší v matrixových neuronech než ve striozomálních neuronech a vyšší v projekčních neuronech než v interneuronech. Selektivní citlivost neuronů tedy koreluje s jejich obsahem huntingtinu, který je normálně přítomen v určitých neuronálních populacích.
Stejně jako v mozku pacientů s Huntingtonovou chorobou, i u myší transgenních pro N-terminální fragment genu Huntingtonovy choroby se zvýšeným počtem repetic, huntingtin tvoří husté agregáty v jádrech neuronů. Tyto intranukleární inkluze se tvoří v neuronech striatální projekce (ale ne v interneuronech). U transgenních myší se inkluze tvoří několik týdnů před nástupem příznaků. Tato data naznačují, že protein huntingtin obsahující zvýšený počet glutaminových zbytků, jejichž inkluze kódují trinukleotidové repeticky, nebo jeho fragment, se hromadí v jádře a může následně narušit jeho kontrolu buněčných funkcí.
Příznaky Huntingtonovy choroby
Věk, ve kterém se u pacientů s Huntingtonovou chorobou objevily první příznaky, je obtížné přesně určit, protože se onemocnění projevuje postupně. Změny osobnosti a chování, mírné poruchy koordinace se mohou objevit mnoho let před objevením se zřetelnějších příznaků. V době stanovení diagnózy má většina pacientů choreické pohyby, zhoršenou koordinaci jemných pohybů a pomalou tvorbu volních sakád. S postupem onemocnění se zhoršuje schopnost organizovat si činnosti, snižuje se paměť, obtížně se řeší, zhoršují se okulomotorické poruchy a zhoršuje se výkon koordinovaných pohybů. Ačkoli v rané fázi onemocnění nedochází ke změnám ve svalech a držení těla, s postupem se mohou vyvinout dystonické držení těla, které se časem mohou stát dominantním příznakem. V pozdní fázi se řeč stává nezřetelnou, polykání se stává výrazně obtížným, chůze se stává nemožnou. Huntingtonova choroba obvykle postupuje 15–20 let. V terminálním stádiu je pacient bezmocný a vyžaduje neustálou péči. Fatální výsledek přímo nesouvisí s primárním onemocněním, ale s jeho komplikacemi, například se zápalem plic.
Demence u Huntingtonovy choroby
Kód MKN-10
P02.2. Demence u Huntingtonovy choroby (G10).
Demence se vyvíjí jako jeden z projevů systémového degenerativně-atrofického procesu s převažujícím poškozením striatálního systému mozku a dalších subcekálních jader. Dědí se autozomálně dominantním způsobem.
Onemocnění se zpravidla projevuje ve třetí nebo čtvrté dekádě života choreoformní hyperkinézou (zejména v obličeji, pažích, ramenou, chůzi), změnami osobnosti (excitovatelné, hysterické a schizoidní typy osobnostních anomálií), psychotickými poruchami (zvláštní deprese s melancholií, zachmuřeností, dysforií; paranoidní náladou).
Pro diagnostiku je obzvláště důležitá kombinace choreoformní hyperkinézy, demence a dědičné zátěže. Pro tuto demenci je specifické následující:
- pomalý postup (v průměru 10–15 let): disociace mezi zbývající schopností postarat se o sebe a zjevnou intelektuální neschopností v situacích vyžadujících produktivní duševní práci (koncepční myšlení, učení se novým věcem);
- výrazná nerovnoměrnost duševní výkonnosti, která je založena na hrubých poruchách pozornosti a nestálosti postojů pacienta („trhavé“ myšlení, podobné hyperkinéze);
- atypičnost zjevných porušení vyšších kortikálních funkcí;
- inverzní vztah mezi nárůstem demence a závažností psychotických poruch.
Vzhledem k vysokému podílu psychotických (paranoidní bludy žárlivosti, pronásledování) a dysforických poruch v klinickém obrazu onemocnění se léčba provádí pomocí různých neuroleptik, která blokují dopaminergní receptory (deriváty fenothiazinu a butyrofenonu) nebo snižují hladinu dopaminu v tkáních (reserpin).
Používá se haloperidol (2-20 mg/den), tiaprid (100-600 mg/den) po dobu maximálně tří měsíců, thioridazin (do 100 mg/den), reserpin (0,25-2 mg/den) a antikonvulzivum klonazepam (1,5-6 mg/den). Tyto léky pomáhají snižovat hyperkinezi, vyhlazovat afektivní napětí a kompenzovat poruchy osobnosti.
Lůžková léčba duševních poruch se provádí s ohledem na vedoucí syndrom, věk a celkový stav pacienta. V ambulantní léčbě jsou principy terapie stejné (kontinuální udržovací terapie poruch hybnosti, periodická změna léku). V ambulantní léčbě se používají nižší dávky neuroleptik.
Rehabilitační opatření pro lehkou a středně těžkou demenci zahrnují ergoterapii, psychoterapii a kognitivní trénink. Je nutné spolupracovat s rodinnými příslušníky a poskytovat psychologickou podporu osobám pečujícím o pacienta. Hlavní metodou prevence onemocnění je lékařské a genetické poradenství nejbližších příbuzných pacienta s doporučením k analýze DNA při rozhodování o těhotenství.
Prognóza je obecně nepříznivá. Průběh onemocnění je pomalu progresivní a obvykle vede k úmrtí po 10–15 letech.
[ 18 ]
Co tě trápí?
Diagnóza Huntingtonovy choroby
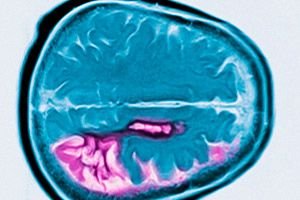
Diagnóza je založena na typických příznacích, rodinné anamnéze a genetickém testování. V důsledku atrofie hlavy nucleus caudatus (jádra caudatus) odhalí MRI a CG v pozdním stádiu onemocnění zvětšení mozkových komor.
Léčba Huntingtonovy choroby
Léčba Huntingtonovy choroby je symptomatická. Choreu a agitovanost lze částečně potlačit neuroleptiky (např. chlorpromazin 25-300 mg perorálně 3krát denně, haloperidol 5-45 mg perorálně 2krát denně) nebo reserpinem 0,1 mg perorálně jednou denně. Dávky se zvyšují na maximum tolerované dávky (než se objeví nežádoucí účinky, jako je ospalost, parkinsonismus; u reserpinu hypotenze). Cílem empirické terapie je snížit glutamatergní přenos přes N-methyl-O-aspartátové receptory a udržet produkci energie v mitochondriích. Léčba zaměřená na zvýšení GABA v mozku je neúčinná.
Genetické testování a poradenství jsou důležité, protože příznaky onemocnění se objevují až po dosažení věku, kdy dítě otěhotní. Lidé s pozitivní rodinnou anamnézou a zájemci o testování jsou s přihlédnutím ke všem etickým a psychologickým důsledkům odesíláni do specializovaných center.
Symptomatická léčba Huntingtonovy choroby
Neexistuje účinná léčba, která by mohla zastavit progresi Huntingtonovy choroby. Bylo provedeno několik studií s různými léky, ale nebylo dosaženo žádného významného účinku. Neuroleptika a další antagonisté dopaminových receptorů se široce používají k nápravě duševních poruch a mimovolních pohybů u pacientů s Huntingtonovou chorobou. Mimovolní pohyby odrážejí nerovnováhu mezi dopaminergním a GABAergním systémem. Neuroleptika se proto používají ke snížení nadměrné dopaminergní aktivity. Tyto léky však samy o sobě mohou způsobovat významné kognitivní a extrapyramidové vedlejší účinky. Kromě toho, s výjimkou případů, kdy se u pacienta rozvine psychóza nebo agitovanost, nebyla jejich účinnost prokázána. Neuroleptika často způsobují nebo zhoršují dysfagii nebo jiné poruchy pohybu. Neuroleptika novější generace, jako je risperidon, klozapin a olanzapin, mohou být obzvláště užitečná při léčbě Huntingtonovy choroby, protože způsobují méně extrapyramidových vedlejších účinků, ale mohou snižovat paranoidní příznaky nebo zvýšenou podrážděnost.
Tetrabenazin a reserpin také snižují aktivitu dopaminergního systému a mohou v raných stádiích onemocnění zmírnit závažnost mimovolních pohybů. Tyto léky však mohou způsobovat depresi. Vzhledem k tomu, že samotné onemocnění často způsobuje depresi, tento vedlejší účinek významně omezuje použití reserpinu a tetrabenazinu. V pozdních stádiích onemocnění buňky nesoucí dopaminové receptory odumírají, takže účinnost antagonistů dopaminových receptorů je oslabena nebo ztracena.
Neuroleptika, antidepresiva a anxiolytika se u pacientů s Huntingtonovou chorobou používají k léčbě psychóz, deprese a podrážděnosti, ale měly by být předepisovány pouze po dobu, kdy pacient tyto příznaky skutečně trpí. Léky, které mohou být užitečné v jedné fázi onemocnění, se mohou s postupem onemocnění stát neúčinnými nebo dokonce škodlivými.
Agonisté receptorů GABA byli testováni u pacientů s Huntingtonovou chorobou, jelikož u Huntingtonovy choroby bylo prokázáno významné snížení hladin GABA ve striatu a také přecitlivělost receptorů GABA v jeho projekčních oblastech. Benzodiazepiny se ukázaly jako účinné v případech, kdy jsou mimovolní pohyby a kognitivní poruchy zhoršeny stresem a úzkostí. Aby se předešlo nežádoucím sedativním účinkům, měly by být předepsány nízké dávky těchto léků. U většiny pacientů s Huntingtonovou chorobou žádný z léků nevede k významnému zlepšení kvality života.
U pacientů s časným nástupem Huntingtonovy choroby s parkinsonskými příznaky lze vyzkoušet dopaminergní látky, ale jejich účinnost je omezená. Levodopa může u těchto pacientů navíc způsobit nebo zesílit myoklonus. Zároveň může baklofen u některých pacientů s Huntingtonovou chorobou snižovat rigiditu.
[ 26 ], [ 27 ], [ 28 ], [ 29 ]
Preventivní (neuroprotektivní) léčba Huntingtonovy choroby
Ačkoli je genetická vada u Huntingtonovy choroby známá, jak vede k selektivní neuronální degeneraci, zůstává nejasné. Předpokládá se, že preventivní terapie zaměřené na snížení oxidačního stresu a excitotoxicity mohou potenciálně zpomalit nebo zastavit progresi onemocnění. Situace může být do jisté míry podobná hepatolentikulární degeneraci, u které genetická vada zůstala po mnoho let neznámá, ale preventivní terapie zaměřené na sekundární efekt, akumulaci mědi, byly „vyléčeny“. V tomto ohledu přitahuje zvláštní pozornost hypotéza, že Huntingtonova choroba je spojena s poruchou energetického metabolismu a buněčnou smrtí v důsledku excitotoxicity. Samotná nemoc může způsobit buněčnou smrt v důsledku intranukleární agregace N-terminálních fragmentů huntingtinu, která narušuje buněčné a metabolické funkce. Tento proces může některé skupiny neuronů postihnout ve větší míře než jiné kvůli jejich vyšší citlivosti na excitotoxické poškození. V tomto případě bude preventivní terapie antagonisty receptorů excitačních aminokyselin nebo látkami, které zabraňují poškození volnými radikály, schopna zabránit vzniku a progresi onemocnění nebo je oddálit. V laboratorních modelech amyotrofické laterální sklerózy bylo prokázáno, že antioxidační látky a antagonisté receptorů (RAA) jsou schopny zpomalit progresi onemocnění. Podobné přístupy mohou být účinné u Huntingtonovy choroby. V současné době probíhají klinické studie antagonistů glutamátových receptorů a látek, které zvyšují funkci komplexu II mitochondriálního elektronového transportního řetězce.
[ 30 ], [ 31 ], [ 32 ], [ 33 ], [ 34 ], [ 35 ], [ 36 ], [ 37 ], [ 38 ], [ 39 ]