
Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
Waldenströmův B-buněčný lymfoplazmocytární lymfom
Lékařský expert článku

Naposledy posuzováno: 12.07.2025
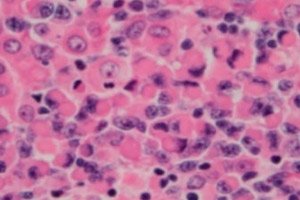
Maligní lymfoproliferativní (imunoproliferativní) onemocnění, lymfoplazmocytární lymfom nebo Waldenstromova makroglobulinemie, je buněčný nádor malých B-lymfocytů – B-buněk, které zajišťují ochranné funkce lymfatického systému a humorální imunitu těla. Diagnóza by měla být stanovena až po vyloučení všech ostatních malých B-buněčných lymfomů. Waldenstromova makroglobulinemie byla popsána v roce 1944 Janem G. Waldenstromem, který u dvou pacientů zaznamenal neobvyklé projevy lymfadenopatického krvácení, anémii, zvýšenou sedimentaci, hyperviskozitu a hypergamaglobulinemii. [ 1 ], [ 2 ]
Epidemiologie
Tento typ lymfomu je vzácný, pomalu se rozvíjející hematologický maligní nádor a klinické statistiky odhadují jeho míru detekce v této skupině onemocnění na přibližně 2 %. Navíc je mezi pacienty mužského pohlaví téměř dvakrát více než žen.
Podle některých údajů je výskyt lymfoplazmocytárního lymfomu ročně v evropských zemích jeden ze 102 tisíc lidí a v USA jeden z 260 tisíc. [ 3 ]
Příčiny lymfoplazmocytární lymfom
Etiologie většiny onkologických onemocnění dosud není známa, ale výzkum genetického základu některých z nich pokračuje. Při studiu příčin maligních onemocnění plazmatických buněk, včetně B-buněčného lymfoplazmocytárního lymfomu – Waldenstromovy makroglobulinémie, vědci objevili souvislost mezi patologickou proliferací (buněčným dělením) B- lymfocytů v pozdní fázi jejich diferenciace s přítomností určitých molekulárně-genových poruch, které mění základní buněčné funkce.
U pacientů s Waldenstromovou makroglobulinemií byly identifikovány změny v některých genech - somatické mutace, tj. postihující pouze tkáně s poškozením genů samostatné klonální populace buněk a tvořící varianty jejich genomu, které vedou k cyklickým a strukturálním poruchám na buněčné úrovni.
V první řadě se jedná o somatické mutace genu MYD88 (L265P) a CXCR4, kódujícího cytosolický protein důležitý pro vrozenou a adaptivní imunitní odpověď: jako adaptér zajišťuje přenos signálů z prozánětlivého mediátoru IL-1 (interleukin-1) a buněk Toll-like receptoru, které aktivují imunitní odpověď. V důsledku somatické mutace vznikají anomálie v polypeptidovém řetězci molekuly tohoto proteinu – jeho strukturním základu. [ 4 ]
Rizikové faktory
Kromě obecných rizikových faktorů (vystavení zvýšeným hladinám radiace, karcinogenním chemikáliím atd.) jsou za prediktory zvýšené pravděpodobnosti vzniku Waldenstromovy makroglobulinémie jakožto lymfoproliferativního onemocnění nízkého stupně považovány následující faktory:
- stáří (nad 65 let);
- přítomnost příbuzných s touto diagnózou, stejně jako s B-buněčným non-Hodgkinovým lymfomem nebo chronickou lymfocytární leukémií;
- chronická hepatitida C;
- anamnéza benigní monoklonální gamapatie, idiopatického hematologického onemocnění, jehož podstatou je produkce abnormálně změněných gama globulinů typu M lymfocytárními plazmatickými buňkami;
- autoimunitní onemocnění, zejména Sjögrenův syndrom.
Patogeneze
Po kontaktu s antigenem nebo stimulaci T-lymfocyty se některé B-lymfocyty transformují na plazmatické buňky – lymfocytární plazmatické buňky, které po určitých transformacích začnou produkovat ochranné globulární proteiny, tj. gama globuliny (imunoglobuliny nebo protilátky).
Patogeneze lymfoplazmocytárního lymfomu/Waldenstromovy makroglobulinémie zahrnuje hyperproliferaci B buněk, nadbytek klonu lymfocytárních plazmatických buněk a nadměrnou produkci imunoglobulinu M (IgM), nazývaného také monoklonální imunoglobulin nebo M protein, v krvi. Jedná se o hlavní protilátku s velkou molekulovou hmotností a pentamerní strukturou, která vzniká během počátečního útoku na specifické bakteriální nebo virové antigeny. [ 5 ]
Téměř všechny příznaky tohoto onemocnění jsou spojeny s projevy aktivity M-proteinu, který může narušit reologické vlastnosti krve, zvýšit její viskozitu; proniknout do lymfoidních a myeloidních tkání kostní dřeně, hromadit se v periferních lymfoidních tkáních (za vzniku pomalu rostoucích novotvarů schopných vyvíjet tlak na okolní orgány, nervová vlákna nebo cévy).
Ačkoli chronická lymfocytární leukémie, Waldenstromova makroglobulinemie nebo lymfoplazmocytární lymfom a mnohočetný myelom jsou samostatná onemocnění, všechna zahrnují zvýšenou proliferaci B lymfocytů.
Symptomy lymfoplazmocytární lymfom
První příznaky onemocnění jsou nespecifické a mohou zahrnovat slabost a zvýšenou únavu (v důsledku rozvoje normochromní anémie), úbytek hmotnosti, dušnost, noční hyperhidrózu a opakující se subfebrilní horečku.
Kromě toho v počáteční fázi onemocnění dochází k porušení citlivosti rukou a nohou, objevuje se periferní neuropatie (necitlivost nebo brnění v nohou a chodidlech), objevují se drobná ložisková krvácení kožních kapilár (purpura) a také studená kopřivka (v důsledku tvorby a agregace abnormálních kryoglobulinových proteinů v krevním séru).
Mezi příznaky spojené se syndromem hyperviskozity patří bolesti hlavy a závratě, poškození sítnice a ztráta zraku, tinnitus a ztráta sluchu, křeče, bolesti svalů, vysoký krevní tlak, spontánní krvácení z nosu a krvácení dásní. U žen se může objevit děložní krvácení.
Dále pozorováno: zvětšené lymfatické uzliny (lymfadenopatie); zvětšená slezina (splenomegalie); srdeční selhání s kardialgií a poruchami srdečního rytmu. Ačkoli je viscerální infiltrace vzácná, může být postižen žaludek a střeva s rozvojem průjmu (často s tučnou stolicí). [ 6 ], [ 7 ]
Formuláře
Klasifikace nádorů hematopoetických a lymfoidních tkání Světové zdravotnické organizace z roku 2017 stanoví čtyři diagnostická kritéria pro Waldenstromovu makroglobulinémii, včetně:
- Přítomnost monoklonální IgM gamapatie
- Infiltrace kostní dřeně malými lymfocyty vykazujícími plazmacytoidní nebo plazmatobuněčnou diferenciaci
- Infiltrace kostní dřeně s intertrabekulární strukturou
- Imunofenotyp odpovídající Waldenstromově makroglobulinémii, která zahrnuje povrchové IgM+, CD19+, CD20+, CD22+, CD25+, CD27+, FMC7+, variabilní CD5, CD10-, CD23-, CD103- a CD108-
Komplikace a důsledky
Pacienti s lymfoplazmocytárním lymfomem vyvíjejí komplikace a následky ve formě:
- snížená imunita;
- selhání kostní dřeně s narušením jejích hematopoetických funkcí a rozvojem anémie;
- nedostatek takových krevních elementů, jako jsou erytrocyty, leukocyty, krevní destičky;
- léze gastrointestinálního traktu s chronickým průjmem a zhoršenou střevní absorpcí (malabsorpční syndrom);
- zánět stěn cév (komplexní imunitní vaskulitida);
- zvýšená křehkost kostí (osteoporóza);
- zrakové a sluchové poruchy;
- sekundární amyloidóza vnitřních orgánů;
- progrese do paraproteinemické hemoblastózy ve formě mnohočetného myelomu;
- transformace do vysoce maligního typu lymfomu – difúzního velkobuněčného B-lymfomu.
Diagnostika lymfoplazmocytární lymfom
Diagnóza lymfoplazmocytárního lymfomu/Waldenstromovy makroglobulinémie je obvykle obtížná kvůli nedostatku specifických morfologických, imunofenotypových nebo chromozomálních změn. Tento nedostatek činí odlišení tohoto onemocnění od jiných malobuněčných lymfomů vylučujícím faktorem.[ 8 ]
Kromě posouzení stávajících symptomů je k diagnostice lymfoplazmocytárního lymfomu nutné provést celkový a biochemický krevní test, koagulogram, imunoelektroforézu krevních proteinů se stanovením hladiny imunoglobulinu M v krvi a celkový test moči. [ 9 ]
Je nutná biopsie kostní dřeně, pro kterou se provádí punkce kostní dřeně.
Provádí se instrumentální diagnostika: ultrazvuk lymfatických uzlin a sleziny, rentgen kostí, CT hrudníku a břišní dutiny, oftalmoskopie.
Diferenciální diagnostika
Lymfoplazmocytární lymfom je považován za diagnózu vyloučení, proto se diferenciální diagnóza provádí s chronickou lymfocytární leukémií B-buněk, mnohočetným myelomem, folikulárním lymfomem, různými podtypy non-Hodgkinova lymfomu, plazmocytomem, reaktivní plazmocytózou, angiofolikulární lymfoidní hyperplazií (Castlemanova choroba) atd.
Kdo kontaktovat?
Léčba lymfoplazmocytární lymfom
Je třeba mít na paměti, že Waldenstromova makroglobulinemie nebo lymfoplazmocytární lymfom může být po mnoho let asymptomatická a diagnostikována zvýšením hladiny M-proteinu v krvi.
Pokud se neobjeví žádné příznaky, provádí se aktivní sledování s pravidelnými vyšetřeními a testy.
Na základě existujících symptomů a výsledků laboratorních testů se rozhodne o zahájení terapie, která závisí na mnoha faktorech (např. věk, progrese onemocnění atd.).
Podle protokolu je počáteční léčba pacientů s tímto typem lymfomu obvykle kombinací radioterapie a chemoterapie s podáváním cytostatik, jako je cyklofosfamid, doxorubicin, vinkristin, a také kortikosteroidů - metprednisolon nebo dexamethason (Dexasone).
Účinnost chemoterapie léky ze skupiny monoklonálních protilátek, zejména rituximabem, byla prokázána. [ 10 ]
V případech generalizovaného onemocnění se Rituximab používá v kombinaci s protinádorovými nukleosidovými analogy (pentostatin, kladribin). U pomalu progredujícího onemocnění s nízkými hladinami monoklonálního imunoglobulinu M se kromě Rituximabu používá cytostatikum Chlorambucil (Leukeran). [ 11 ]
Pro snížení viskozity krve a stabilizaci hladiny jejích vytvořených prvků se používá terapeutická hemaferéza.
Pokud je hladina protilátek v krvi kriticky nízká, provádí se substituční terapie imunoglobuliny, aby se zabránilo souběžným opakovaným infekcím.
Jak poznamenávají onkohematologové, i přes skutečnost, že léčba může vést k remisi onemocnění, u většiny pacientů dochází k relapsu. Pokud k němu dojde dříve než za 24 měsíců, lze použít protinádorový lék, jako je Ibrutinib (ve formě tablet). V případě pozdějších relapsů se léčba provádí podle původního schématu. [ 12 ], [ 13 ], [ 14 ]
Prevence
Odborníci určují prognózu výsledku lymfoplazmocytárního lymfomu podle mezinárodního prognostického systému pro posouzení hlavních parametrů: věku pacienta a sérových hladin hemoglobinu, krevních destiček, beta-2-mikroglobulinu a monoklonálního imunoglobulinu. [ 15 ], [ 16 ]
Průměrná míra přežití u této diagnózy je asi pět let, ale téměř 40 % pacientů žije deset let nebo déle.