
Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
Lissencefalie mozku
Lékařský expert článku

Naposledy posuzováno: 12.07.2025
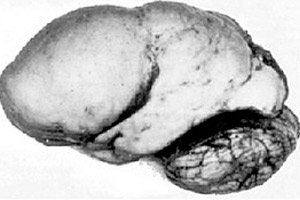
Mezi organickými mozkovými patologií vyniká vrozená anomálie vývoje mozku, jako je lissencefalie, jejíž podstata spočívá v téměř hladkém povrchu kůry jejích hemisfér - s nedostatečným počtem závitů a rýh. [ 1 ]
Při úplné absenci závitů se definuje agyrie a přítomnost několika širokých plochých závitů se nazývá pachygyrie. Tyto vady, stejně jako některé další redukční deformace mozku, mají v MKN-10 kód Q04.3.
Epidemiologie
Podle statistik o vzácných onemocněních připadá 1–1,2 případu lissencefalie na 100 tisíc novorozenců. [ 2 ], [ 3 ]
Podle některých údajů je u dětí s Millerovým-Diekerovým syndromem pozorováno až 25–30 % případů klasické lissencefalie; bodové mutace a delece genů LIS1 a DCX jsou detekovány u téměř 85 % pacientů. [ 4 ]
Genetické studie 17 genů spojených s lissencefalií ukázaly, že mutace nebo delece LIS1 je příčinou 40 % pacientů a 23 % je spojeno s mutací DCX, následovanou TUBA1A (5 %) a DYNC1H1 (3 %).[ 5 ]
Příčiny lissencefalie
Všechny známé příčiny vzniku mozkové kůry (cortex cerebri) téměř nebo úplně bez závitů a rýh, které zvětšují „pracovní plochu“ lidského mozku a zajišťují „produktivitu“ centrálního nervového systému, jsou spojeny s poruchami jeho perinatálního vývoje. To znamená, že u plodu se vyvíjí lissencefalie. [ 6 ]
Neschopnost tvořit vrstvy mozkové kůry plodu u lisencefalie je výsledkem abnormální migrace neuronů, které ji tvoří, nebo předčasného ukončení tohoto procesu.
Tento proces, který je nezbytný pro cerebrokortikální histogenezi, probíhá v několika fázích od 7. do 18. týdne těhotenství. A vzhledem k jeho zvýšené citlivosti na genetické mutace, stejně jako na různé negativní fyzikální, chemické a biologické vlivy, může jakákoli odchylka od normy vést k nesprávné lokalizaci neuronů s možnou tvorbou ztluštělé vrstvy šedé hmoty kůry bez charakteristické struktury. [ 7 ]
V některých případech je lissencefalie u dětí spojena s Millerovým-Diekerovým, Walkerovým-Warburgovým nebo Normanovým-Robertsovým syndromem.
Čtěte také – Vývojové vady mozku
Rizikové faktory
Kromě mutací v některých genech patří mezi rizikové faktory pro narození dítěte s tak závažnou vadou kyslíkové hladovění (hypoxie) plodu; nedostatečné prokrvení mozku (hypoperfúze); akutní cévní mozková příhoda ve formě perinatální mrtvice; patologie placenty; virové infekce těhotné ženy (včetně TORCH); [ 8 ] problémy s celkovým metabolismem a funkcí štítné žlázy; kouření, alkohol, psychotropní a omamné látky; užívání řady léků; zvýšené hladiny radiace. [ 9 ]
Patogeneze
Ne všechny případy lissencefalie mají patogenezi způsobenou chromozomálními abnormalitami a genovými mutacemi. Jsou však známy některé geny, které kódují proteiny hrající důležitou roli ve správném pohybu neuroblastů a neuronů podél radiálních gliových buněk – za vzniku mozkové kůry. A mutace těchto genů vedou k této patologii. [ 10 ]
Zejména se jedná o sporadické mutace (bez dědičnosti) genu LIS1 na chromozomu 17, který reguluje cytoplazmatický motorický protein mikrotubulů dynein, a také gen DCX na chromozomu X, který kóduje protein doublecortin (lisencefalin-X). [ 11 ]. V prvním případě specialisté definují klasickou lissencefalii (typ I), ve druhém – vázanou na chromozom X. [ 12 ]
Pokud je gen FLN1, který kóduje fosfoprotein filamin 1, deletován, proces řízené neuronální migrace nemusí vůbec začít, což vede k úplné absenci konvolucí (agyrie). [ 13 ]
Byly identifikovány mutace v genu CDK5, který kóduje kinázový enzym – katalyzátor intracelulárního metabolismu, regulující buněčný cyklus v neuronech CNS a zajišťující jejich normální migraci během prenatálního formování mozkových struktur.
Abnormální změny v genu RELN na chromozomu 7, které způsobují kortikální gyrální defekty u Norman-Robertsova syndromu, vedou k deficitu extracelulárního glykoproteinu reelinu, který je nezbytný pro regulaci migrace a umístění nervových kmenových buněk během vývoje mozkové kůry.[ 14 ], [ 15 ], [ 16 ]
Gen ARX kóduje non-aristalens homeobox protein, transkripční faktor, který hraje důležitou roli v předním mozku a dalších tkáních.[ 17 ] Děti s mutací ARX mají další příznaky, jako jsou chybějící části mozku (ageneze corpus callosum), abnormální genitálie a těžká epilepsie.[ 18 ],[ 19 ]
S lissencefalií bylo spojeno několik genů. Mezi tyto geny patří VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A a CDK5.[ 20 ]
Cytomegalovirus (CMV) je spojován s rozvojem lissencefalie v důsledku sníženého prokrvení mozku plodu. Závažnost CMV infekce závisí na gestačním věku. Časná infekce s větší pravděpodobností způsobí lissencefalii, protože k migraci neuronů dochází v raném těhotenství.[ 21 ]
Mechanismus výskytu této anomálie navíc zahrnuje neúplné nebo pozdější zastavení migrace neuronů z periventrikulární generativní zóny do mozkové kůry. A v takových případech se vyvíjí buď neúplná lissencefalie, nebo pachygyrie, ve které se tvoří několik širokých drážek a závitů (ale většina z nich chybí).
Symptomy lissencefalie
První příznaky této patologie (při absenci dříve zmíněných syndromů) se nemusí objevit ihned po narození, ale po jednom a půl až dvou měsících. A nejčastěji se pozorují následující klinické příznaky lissencefalie:
- svalová hypotonie, často kombinovaná se spastickou paralýzou;
- křeče a generalizované tonicko-klonické záchvaty (ve formě opisthotonu);
- těžká mentální retardace a růstová retardace;
- zhoršení neurologických a motorických funkcí.
Problémy s polykáním způsobují potíže s krmením dítěte. [ 22 ]
Vysoký stupeň neuromotorického postižení se často projevuje tetraplegií – paralýzou všech končetin. Možná je deformace rukou, prstů na rukou nebo nohou.
U Norman-Robertsova syndromu s lissencefalií typu I se pozorují kraniofaciální anomálie: těžká mikrocefalie, nízký sklon čela a vyčnívající široký kořen nosu, široce posazené oči (hyperterlorismus), nedostatečný vývoj čelistí (mikrognatie). [ 23 ]
Millerův-Diekerův syndrom může být také charakterizován abnormálně malou velikostí hlavy se širokým, vysokým čelem a krátkým nosem, prohlubněmi v spáncích (bitemporálními prohlubněmi) a nízko posazenými, deformovanými ušima.
Syndrom těžké lissencefalie je charakterizován mikrocefalií, zmenšením velikosti očních bulv (mikroftalmie), v kombinaci s retinální dysplazií, obstrukčním hydrocefalem a chybějícím nebo hypoplastickým corpus callosum.
Komplikace a důsledky
Mezi komplikace této anomálie odborníci uvádějí poruchu polykací funkce (dysfagie) a gastroezofageální reflux; refrakterní (nekontrolovanou) epilepsii; časté infekce horních cest dýchacích; pneumonii (včetně chronické aspirace).
Kojenci s lissencefalií mohou mít vrozené organické srdeční problémy ve formě defektu síňového septa nebo komplexní srdeční vady s cyanózou (Fallotova tetralogie). [ 24 ]
Důsledky postnatální růstové retardace ve většině případů vedou k úmrtí do 24 měsíců po narození.
Diagnostika lissencefalie
Diagnóza začíná fyzikálním vyšetřením dítěte, studiem anamnézy rodičů a anamnézy těhotenství a porodu.
Během těhotenství může být nutné provést testování fetální DNA bez buněk, amniocentézu nebo odběr vzorků choriových klků. [ 25 ] Více informací viz – Prenatální diagnostika vrozených onemocnění
Instrumentální diagnostika se používá k vizualizaci mozkových struktur a posouzení jejich funkcí:
- počítačová tomografie mozku;
- magnetická rezonance (MRI) mozku;
- elektroencefalogram (EEG). [ 26 ]
Během těhotenství lze na ultrazvuku plodu po 20-21 týdnech podezřívat lissencefálii při absenci parietookcipitálních a kalkarálních rýh a anomálii Sylvianské štěrbiny mozku.
Diferenciální diagnostika
Provádí se diferenciální diagnostika s jinými syndromy vrozených mozkových vad.
Existuje více než 20 typů lissencefalie, z nichž většina spadá do 2 hlavních kategorií: klasická lissencefalie (typ 1) a lissencefalie typu cobblestone (typ 2). Každá kategorie má podobné klinické projevy, ale odlišné genetické mutace.[ 27 ]
Vyšetření mozku u lisencefalie typu I ukazuje mozkovou kůru se čtyřmi vrstvami namísto šesti, jak je vidět u normálních pacientů, zatímco u lisencefalie typu 2 je mozková kůra dezorganizovaná a jeví se hrudkovitá nebo nodulární v důsledku úplného posunutí mozkové kůry shluky kortikálních neuronů oddělených gliomesenchymální tkání. Pacienti měli také svalové a oční abnormality.
- Klasická lissencefalie (typ 1):
- LIS1: Izolovaná lissencefalie a Millerův-Diekerův syndrom (lissencefalie spojená s dysmorfismem obličeje). [ 28 ]
- LISX1: Mutace genu DCX. Ve srovnání s lissencefalií způsobenou mutacemi LIS1 vykazuje DCX šestivrstvou kůru místo čtyř.
- Izolovaná lissencefalie bez dalších známých genetických vad
- Dlažební kostková lissencefalie (typ 2):
- Walkerův-Warburgův syndrom
- Fukuyamův syndrom
- Onemocnění svalů, očí a mozku
- Jiné typy nelze zařadit do žádné z výše uvedených dvou skupin:
- LIS2: Normanův-Robertsův syndrom, podobný lissencefalii typu I nebo Millerovu-Diekerovu syndromu, ale bez delece chromozomu 17.
- LIS3
- LISX2
Mikrolisencefalie: Jedná se o kombinaci absence normálního kortikálního záhybu a abnormálně malé hlavy. Děti s pravidelnou lissencefalií mají při narození normální velikost hlavy. Děti s malou velikostí hlavy při narození jsou obvykle diagnostikovány s mikrolisencefalií.
Je také důležité rozlišovat mezi lissencefalií a polymikrogyrií, což jsou různé vývojové vady mozku.
Kdo kontaktovat?
Léčba lissencefalie
Lissencefalie je nevyléčitelná organická vada, takže je možná pouze podpůrná a symptomatická léčba. [ 29 ]
V první řadě se jedná o užívání antikonvulziv a antiepileptik, stejně jako o zavedení gastrostomické sondy do žaludku (pokud dítě není schopno samostatně polykat). Masáž je užitečná.
V případech těžkého hydrocefalu se odstraňuje mozkomíšní mok.
Prevence
Odborníci doporučují, aby budoucí rodiče vyhledali genetické poradenství a těhotné ženy se včas zaregistrovaly u porodníků a gynekologů a podstoupily všechna plánovaná vyšetření.
Předpověď
U dětí s lissencefalií závisí prognóza na jejím stupni, ale nejčastěji mentální vývoj dítěte nepřesahuje úroveň čtyř až pěti měsíců. A všechny děti s touto diagnózou trpí těžkými psychomotorickými poruchami a obtížně léčitelnou epilepsií. [ 30 ]
Podle NINDS (Americký národní institut neurologických poruch a mrtvice) je maximální délka života lidí s lissencefalií přibližně 10 let.