
Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
Angelmanův syndrom u dětí a dospělých
Lékařský expert článku

Naposledy posuzováno: 04.07.2025

Existuje řada nemocí, u kterých výrazy jako „starej se o sebe a neonemocníš“ zní přinejmenším směšně. Jde o patologie, u kterých jsou některé duševní a fyzické abnormality vrozené tělu dítěte ještě před narozením, ale rodiče za to nenesou vinu. Taková onemocnění jsou způsobena mutacemi nebo abnormalitami v chromozomálních sadách a nazývají se chromozomální nebo genetická. Angelmanův syndrom, Downův syndrom, Patauův syndrom, Edwardsův syndrom, Turnerův syndrom, Prader-Williho syndrom – to je jen část genetických onemocnění z poměrně slušného seznamu.
Syndrom šťastného muže
Tentokrát si povíme o patologii pojmenované po anglickém pediatrovi Harrym Angelmanovi, který se na tento problém poprvé zaměřil v roce 1965, když se den předtím ve své praxi setkal se třemi neobvyklými dětmi, které spojovaly společné specifické příznaky. Lékař tyto děti nazval panenským dětstvím a napsal o nich článek, který se původně jmenoval „Děti-loutky“. Samotný článek i jeho název byly napsány pod dojmem obrazu, který byl vidět v jednom z muzeí ve Veroně. Obraz zobrazoval smějícího se chlapce a jmenoval se „Loutkový chlapec“. Spojení dítěte zobrazeného na obraze se třemi dětmi, se kterými se Angelman kdysi ve své praxi setkal, vedlo pediatra ke sloučení dětí do jedné skupiny kvůli jejich onemocnění.
Není nic překvapivého na tom, že si dětí zmíněných v článku ostatní lékaři nevšimli. Vždyť se na první pohled zdálo, že mají zcela odlišná onemocnění, tak odlišný byl celkový klinický obraz onemocnění ve 3 různých případech. Možná by „nová“ chromozomální patologie zaujala i jiné vědce, ale v té době genetika ještě nebyla dostatečně rozvinutá, aby potvrdila hypotézu anglického lékaře. Proto byl článek po určitém zájmu o něj na dlouhou dobu odložen na zadní poličku.
Další zmínka o Angelmanově syndromu, jak se nyní nazýval článek anglického pediatra G. Angelmana, pochází z počátku 80. let 20. století. A teprve v roce 1987 se podařilo najít důvod, proč se malá část dětí rodí s takovými odchylkami, že se navenek zdají být neustále usměvavé a šťastné. Ve skutečnosti to vůbec není pravda a úsměv je jen grimasa, za kterou se skrývá nešťastná lidská duše a bolest rodičů.
Epidemiologie
Podle statistik se chromozomální mutace u dítěte může vyvinout jak na pozadí podobných mutací u rodičů, tak i v jejich nepřítomnosti. Neexistuje jasná dědičná povaha Angelmanova syndromu (AS), ale pravděpodobnost vzniku patologie u rodičů s chromozomálními mutacemi je poměrně vysoká.
Zajímavé je také, že pokud rodina již má dítě s AS, existuje jednoprocentní šance, že se narodí druhé dítě se stejnou poruchou, a to i v případě, že jsou rodiče zdraví.
Stále neexistují přesné statistiky o počtu pacientů s Angelmanovým syndromem. Důvodem je možná rozmanitost symptomů, které se mohou vyskytovat v určitém složení nebo se po dlouhou dobu vůbec neobjevovat. Předpokládá se, že prevalence onemocnění je: 1 dítě na 20 000 novorozenců. Toto číslo je však velmi přibližné.
Příčiny Angelmanův syndrom
Angelmanův syndrom je lékařský název pro chromozomální patologii, ale zdaleka není jediný. Lidé tuto nemoc nazývají syndromem panenek-dětí, syndromem šťastné loutky, syndromem Petrušky a syndromem smějící se panenky. Lidé vymýšlejí nejrůznější názvy (někdy i urážlivé pro samotné pacienty a jejich rodiče), ale nemoc je nemoc, bez ohledu na to, jak legrační může vypadat a bez ohledu na to, jaké jsou její důvody.
A důvody pro rozvoj Angelmanova syndromu, stejně jako mnoho jiných genetických patologií, jsou ve všech případech poruchy ve struktuře jednoho z chromozomů nebo chromozomální sady jako celku. Ale v našem případě spočívá celý problém v chromozomu 15, předávaném od matky. To znamená, že otcovský chromozom v tomto případě nemá žádné odchylky, ale ženský prochází určitými mutacemi.
Podle typu chromozomální abnormality je Angelmanův syndrom klasifikován jako chromozomální mutace. Za takové mutace se považují:
- Delece (absence úseku chromozomu obsahujícího určitou sadu genů; pokud jeden z genů chybí, mluvíme o mikrodeleci), která je výsledkem dvou zlomů a jednoho sloučení, kdy dochází ke ztrátě úseku původního chromozomu.
- Duplikace (přítomnost další části chromozomu, která je kopií existující části), která ve většině případů vede k úmrtí osoby a méně často k neplodnosti.
- Inverze (otočení jednoho z úseků chromozomu o 180 stupňů, tj. opačným směrem, a geny v něm se pak nacházejí v opačném pořadí), kdy se přerušené konce chromozomu spojí v pořadí odlišném od původního.
- Vložení (pokud je část genetického materiálu v chromozomu na nesprávném místě),
- translokace (pokud je určitá část chromozomu připojena k jinému chromozomu; taková mutace může být vzájemná bez ztráty částí).
Pokud dítě od nic netušící matky obdrží mutovaný chromozom, je odsouzeno k narození s abnormalitami. Za nejčastější příčinu Angelmanova syndromu se stále považuje delece mateřského 15. chromozomu, kdy chybí malá část. Za méně časté mutace u syndromu „smějící se panenky“ se považují:
- translokace,
- unipaternální disomie (pokud dítě zdědilo od otce pár chromozomů, mateřský chromozom chybí),
- mutace genů v DNA, které jsou jak hlavním stavebním (genetickým) materiálem, tak i návodem k jeho správnému použití (zejména mutace genu ube3a v mateřském chromozomu).
Přítomnost jedné z těchto mutací u rodičů je rizikovým faktorem pro rozvoj Angelmanova syndromu u dětí. Nejen chromozomální mutace, ale i genomické (které jsou spojeny s kvantitativní změnou chromozomálních sad a jsou častější než chromozomální) mohou vyvolat rozvoj onemocnění u dítěte. Mezi běžné genomové mutace patří chromozomální trizomie (pokud chromozomální sada osoby má více než 46 chromozomů).
Aby se u dítěte objevila patologie, není vůbec nutné, aby rodiče měli chromozomální abnormality. Přesto existuje určité procento pacientů, jejichž onemocnění je dědičné.
Patogeneze
Pojďme se trochu hlouběji ponořit do biologie, přesněji řečeno do genetiky. Genetická informace každého jednotlivého lidského organismu je obsažena ve 23 párech chromozomů. Jeden chromozom z páru se na dítě předává od otce, druhý od matky. Všechny páry chromozomů se liší tvarem a velikostí a nesou určitou informaci. 23. pár chromozomů (chromozomy X a Y) je tedy zodpovědný za formování pohlavních znaků dítěte (XX - dívka, XY - chlapec, zatímco chromozom Y může dítě obdržet pouze od otce).
V ideálním případě dítě od rodičů dostane 46 chromozomů, které tvoří jeho genetické vlastnosti a předurčují ho jako jedince. Větší počet chromozomů se nazývá trizomie a je považován za odchylku od normy. Například přítomnost chromozomu 47 v chromozomální sadě (karyotyp, určující druh a individuální vlastnosti) způsobuje výskyt Downova syndromu.
Pokud jsou chromozomy obarveny speciálním barvivem, pak pod mikroskopem můžete podél každého z nich vidět pruhy různých odstínů. Uvnitř každého pruhu je obrovské množství genů. Všechny tyto pruhy jsou vědci očíslovány a mají pevnou polohu. Absence jednoho z pruhů je považována za odchylku od normy. U Angelmanova syndromu lze velmi často pozorovat absenci segmentů mateřského chromozomu v intervalu q11-q13, umístěných v dlouhém rameni, jehož počet DNA bází je pouze asi 4 miliony.
Hlavní složkou chromozomu je neuvěřitelně dlouhá molekula DNA obsahující tisíce genů a desítky a stovky milionů dusíkatých bází. Chromozom 15, zodpovědný za rozvoj Angelmanova syndromu a několika dalších, tedy obsahuje 1200 genů a asi 100 milionů bází. Jakékoli narušení struktury molekuly DNA jistě ovlivní vzhled a vývoj budoucího dítěte.
Genetická informace obsažená v genech se přeměňuje na protein nebo RNA. Tento proces se nazývá genová exprese. Tímto způsobem genetická informace přijatá od rodičů dostává formu i obsah, který je ztělesněn v jejich jedinečném ženském nebo mužském dědici.
Existuje řada patologií s neklasickým typem dědičnosti, včetně Angelmanova syndromu, u kterého geny přijaté od rodičů jako součást párových chromozomů nesou jedinečný otisk rodičů a projevují se různými způsoby.
Angelmanův syndrom je tedy pozoruhodným příkladem genomického imprintingu, kdy je genová exprese v těle dítěte přímo závislá na tom, od kterého rodiče byly alely přijaty (různé formy jednoho genu, přijaté od otce a matky, umístěné na identických úsecích párových chromozomů). To znamená, že k rozvoji syndromu vedou pouze anomálie v mateřském chromozomu, zatímco mutace a strukturální poruchy otcovského chromozomu způsobují zcela odlišné patologie.
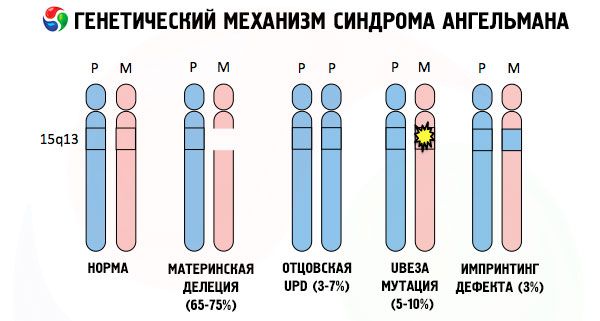
U této patologie dochází k nedostatku určitých genů v mateřském chromozomu nebo ke ztrátě/snížení aktivity jednotlivých genů (ve velké většině případů genu ube3a, který se podílí na metabolismu ubikvitinu, proteinu regulujícího odbourávání jiných proteinů). V důsledku toho je u dítěte diagnostikována mentální vývojová abnormalita a fyzické deformace.
Symptomy Angelmanův syndrom
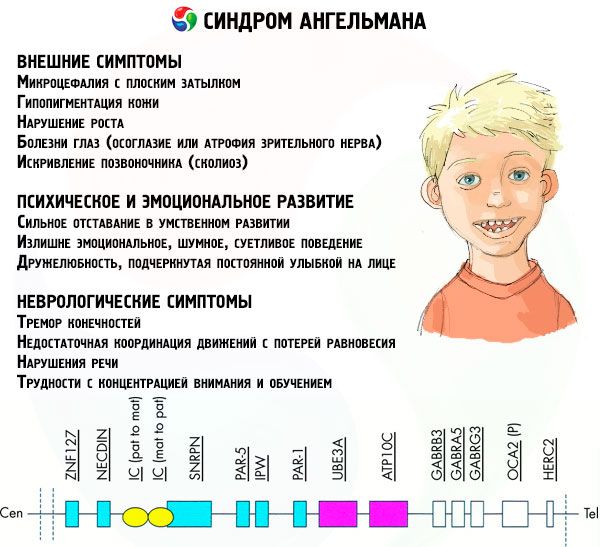
Příznaky Angelmanova syndromu ovlivňují různé aspekty života a vývoje dítěte: fyzické, neurologické, psychické. Na základě toho lze identifikovat 3 skupiny příznaků, které naznačují vývoj této patologie.
- Vnější nebo fyzické příznaky:
- neúměrně malá hlava ve srovnání s tělem a končetinami, které mají normální velikost,
- příliš široká ústa,
- na tváři je téměř vždy úsměv (s otevřenými ústy),
- řídké zuby,
- úzký horní ret,
- často vyplazený široký jazyk,
- vyčnívající spodní čelist,
- špičatá brada,
- velmi světlá kůže, často i vlasy (albinismus, spojený s tím, že tělo neprodukuje pigment melanin),
- tmavé skvrny na světlé pleti (hypopigmentace v důsledku nedostatečné produkce melaninu)
- fyzické nebo vnější příznaky: oční onemocnění, jako je strabismus nebo atrofie zrakového nervu,
- zakřivení páteře (skolióza),
- ztuhlé nohy (při chůzi si člověk neohýbá nohy v kolenou kvůli nízké pohyblivosti kloubů, proto srovnání s chůzí panenky).
- Příznaky související s duševním a emocionálním vývojem:
- těžká mentální retardace,
- přehnaně emocionální, hlučné, rozrušené chování,
- časté tleskání rukou,
- projevená přátelskost, zdůrazněná neustálým úsměvem na tváři,
- častý smích bez důvodu.
- Neurologické příznaky:
- třes končetin,
- nedostatečná koordinace pohybů se ztrátou rovnováhy,
- snížený svalový tonus,
- různé poruchy spánku,
- časté hysterické záchvaty v dětství,
- poruchy řeči (dítě začíná mluvit pozdě, má špatné komunikační dovednosti a nezřetelnou řeč),
- hyperaktivita na pozadí zvýšené excitability,
- potíže s koncentrací a učením.
Jedná se však o zobecněný obraz onemocnění. Ve skutečnosti klinický obraz Angelmanova syndromu do značné míry závisí na stádiu vývoje onemocnění a typu chromozomální mutace, která patologii způsobila. To znamená, že příznaky onemocnění se u různých pacientů mohou výrazně lišit, což nám po dlouhou dobu neumožňovalo odlišit patologii od jiných s podobným klinickým obrazem.
Z celkového počtu symptomů můžeme vyzdvihnout ty, které jsou charakteristické pro všechny pacienty bez výjimky:
- těžká mentální retardace,
- nevhodné chování (bezdůvodný smích, zvýšená podrážděnost, špatná koncentrace, stav euforie),
- nedostatečný rozvoj motorických dovedností,
- špatná koordinace pohybů, ataxie chůze (nerovnoměrné tempo, kymácení ze strany na stranu atd.), třes končetin.
- porucha vývoje řeči s převahou neverbálních komunikačních prostředků.
Mezi příznaky, se kterými se setkává drtivá většina pacientů, lze rozlišit následující:
- nepoměr mezi hlavou a tělem způsobený opožděným fyzickým vývojem,
- u mnoha pacientů je tvar lebky takový, že velikost mozku zůstává menší než u zdravých lidí (mikrocefalie),
- epileptické záchvaty před dosažením věku 3 let s progresivním poklesem síly a frekvence ve vyšším věku,
- zkreslení EEG parametrů (fluktuace a vysoká amplituda nízkofrekvenčních vln).
Tyto příznaky jsou poměrně časté, nicméně 20 % pacientů s Angelmanovým syndromem je nemá.
Ještě méně často je možné diagnostikovat takové projevy onemocnění, jako jsou:
- těžký nebo mírný strabismus,
- špatná kontrola pohybu jazyka, což má za následek, že pacienti často vyplazují jazyk bezdůvodně,
- potíže s polykáním a sáním, zejména u malých dětí,
- narušení pigmentace kůže a očí,
- paže zdvižené nebo pokrčené při chůzi,
- hyperreflexie,
- poruchy spánku, zejména v dětství,
- časté slinění,
- neukojitelná žízeň,
- příliš aktivní žvýkací pohyby,
- přecitlivělost na teplo,
- plochá zadní část hlavy,
- vyčnívající spodní čelist,
- hladké dlaně.
Poměrně velké procento pacientů má problémy s močením, které špatně ovládají, zhoršenou jemnou motoriku, která jim způsobuje potíže s péčí o sebe a učením, a nadváhu. Téměř všichni pacienti prožívají pubertu později než zdraví vrstevníci.
Děti s Angelmanovým syndromem dobře vnímají ústní řeč a rozumí jí, ale nechtějí se účastnit konverzace a omezují svou řeč na několik desítek slov nezbytných v každodenním životě. V dospělosti však tito pacienti vypadají mladší než jejich vrstevníci bez genetických patologií.
Mnoho příznaků Angelmanova syndromu je nestálých, takže klinický obraz onemocnění se s věkem významně mění. Křeče a epileptické záchvaty se stávají méně častými nebo zcela mizí, pacient se stává méně vzrušivým a zlepšuje se spánek.
Komplikace a důsledky
Angelmanův syndrom je závažná, v současnosti prakticky nevyléčitelná chromozomální patologie, která pacientům brání v možnosti žít normální život. Jaký bude život dítěte s AS, do značné míry závisí na typu chromozomální abnormality.
Duplikace segmentu chromozomu je ve většině případů neslučitelná se životem. A i když takoví pacienti nezemřou v kojeneckém věku a dosáhnou puberty, nemají šanci mít děti.
Vymazání nebo absence části genů, která se nejčastěji vyskytuje u Angelmanova syndromu, je překážkou pro dítě, aby se naučilo chodit a mluvit. Takové děti mají těžší formu mentální retardace a epileptické záchvaty se vyskytují častěji a jejich intenzita je mnohem větší než u pacientů s jinými chromozomálními abnormalitami.
Pokud se jedná pouze o mutaci jednoho genu, lze dítě s patřičnou pozorností a přístupem naučit základy sebeobsluhy, komunikace a interakce ve skupině, i když bude ve vývoji stále zaostávat za svými vrstevníky.
Pro děti s Angelmanovým syndromem, které jsou od přírody laskavé, je nejdůležitější láska a pozornost rodičů. Pouze v tomto případě přinese vzdělání dítěte ovoce, byť jen malé. Pacienti s AS samozřejmě nebudou moci studovat v běžné škole. Potřebují speciální třídy, kde se děti nejprve naučí soustředit a poté se jim postupně budou předávat základy školních znalostí.
Diagnostika Angelmanův syndrom
Angelmanův syndrom je vrozená vývojová patologie. Vzhledem k určitým okolnostem je však často nemožné jej diagnostikovat v kojeneckém a raném dětství. To je způsobeno nespecificitou a slabou expresí symptomů u kojenců a dětí mladších 3 let. A prevalence onemocnění v naší zemi není tak velká, aby se lékaři naučili jej rozpoznávat mezi jeho vrstevníky.
Angelmanův syndrom se u kojenců může projevovat sníženým svalovým tonem, který se projevuje problémy s krmením (slabost sacího a polykacího reflexu) a později obtížemi s učením se chůze (takové děti začínají chodit mnohem později). Tyto příznaky jsou prvními známkami vývojové abnormality u dítěte, která může být spojena s chromozomální abnormalitou. Tuto domněnku může potvrdit pouze genetická analýza.
Zvláštní pozornost je věnována dětem, jejichž rodiče mají různé genomové nebo chromozomální poruchy. Onemocnění se totiž zpočátku nemusí projevit a pokud je patologie odhalena včas, je možné zahájením intenzivní práce s dítětem dosáhnout výrazně větších úspěchů v učení a zpomalit tak progresi onemocnění.
Pokud mají rodiče různé chromozomální abnormality, genetická analýza se provádí ještě před narozením dítěte, protože SA je jednou z patologií, které lze detekovat v embryonálním stádiu.
Sběr materiálu pro genetický výzkum lze provádět dvěma způsoby:
- invazivní (s určitým procentem rizika, protože pro odběr vzorku plodové vody je nutné proniknout do dělohy),
- neinvazivní (analýza DNA dítěte z krve matky).
Poté se provedou následující studie:
- fluorescenční in situ hybridizace (metoda FISH) – vazba DNA sondy značené speciálním barvivem na studovanou DNA a následné vyšetření pod mikroskopem.
- analýza mutací v genu ube3a a imprintovaných genech,
- Analýza metylace DNA pomocí speciálních metod používaných v genetice.
Genetické testy poskytují v případě chromozomálních abnormalit poměrně přesné informace, což znamená, že budoucí rodiče předem vědí, na co se mají připravit. Existují však výjimky. U určité skupiny pacientů, pokud jsou přítomny všechny příznaky naznačující patologii, zůstávají výsledky testů normální. To znamená, že patologii lze identifikovat pouze pečlivým pozorováním dítěte od raného dětství: jak jí, kdy začalo chodit a mluvit, zda při chůzi ohýbá nohy atd.
Kromě metody FISH lze mezi instrumentální diagnostické metody Angelmanova syndromu rozlišit tomografii (CT nebo MRI), která pomáhá určit stav a velikost mozku, a elektroencefalogram (EEG), který ukazuje, jak fungují jednotlivé části mozku.
Lékaři obvykle stanoví konečnou diagnózu ve věku 3-7 let, kdy pacient již má většinu příznaků a je viditelná dynamika vývoje onemocnění.
Jaké testy jsou potřeba?
Diferenciální diagnostika
Angelmanův syndrom je genetická patologie, která prakticky nemá žádné specifické projevy. Většina symptomů může stejnou měrou naznačovat jak AS, tak i jiné genetické patologie.
Diferenciální diagnostika Angelmanova syndromu se provádí s následujícími patologií:
- Pitt-Hopkinsův syndrom (pacienti se vyznačují mentální retardací, veselou povahou, úsměvem, mají poměrně velká a široká ústa, je zaznamenána mikrocefalie). Rozdílem jsou záchvaty hyperventilace a zadržování dechu v bdělém stavu.
- Christiansonův syndrom (pacienti jsou mentálně retardovaní lidé s veselou povahou, neschopní mluvit, charakterizovaní mikrocefalií, ataxií, křečemi, mimovolními pohyby svalů).
- Mowat-Wilsonův syndrom (příznaky: mentální retardace, epileptické záchvaty, špičatá brada, otevřená ústa, šťastný výraz ve tváři, mikrocefalie). Rozlišovací znaky: velká vzdálenost mezi očima, oči skloněné dovnitř, zaoblená špička nosu, dozadu otočená ušní boltec.
- Kabukiho syndrom (charakterizovaný mírnou až středně těžkou mentální retardací, řečovými a motorickými problémy, svalovou slabostí, epileptickými záchvaty, mikrocefalií, dlouhými intervaly mezi svěděním a zhoršenou koordinací). Charakteristický je klenutým obočím, vykloubenou laterální částí dolního víčka, široce posazenýma očima, dlouhými očními štěrbinami s dlouhými, hustými řasami.
- Rettův syndrom (rozlišování od AS u žen). Příznaky: opožděný vývoj řeči, záchvaty, mikrocefalie. Rozdíl spočívá v tom, že v obličeji není veselý výraz, objevují se záchvaty apnoe a apraxie, které postupem času progredují.
- Autozomálně recesivní syndrom mentální tardace 38 (příznaky: výrazná mentální retardace se zpožděním motorických dovedností a řeči, svalová slabost, problémy s krmením v kojeneckém věku, impulzivita). Charakteristickým znakem je modrá barva duhovky.
- Syndrom duplikace genu MECP 2 (odlišuje se od SA u mužů). Příznaky: těžká mentální retardace, svalová slabost od dětství, problémy s řečí nebo její absence, epilepsie. Rozlišovací znaky: progresivní myopatie, neustále se opakující infekce.
- Kleefstrův syndrom (příznaky: problémy s řečí a myšlením, svalová slabost, poruchy spánku, nedostatek pozornosti, otevřená ústa, hyperaktivita, záchvaty, ataxie, poruchy rovnováhy). Charakteristické rysy: plochý obličej, krátký tupý nos, široce posazené oči, velký vykloubený spodní ret, agresivní výbuchy.
- Smithův-Magenisův syndrom (charakterizovaný záchvaty, problémy se spánkem, poruchami intelektuálního a motorického vývoje). Mezi charakteristické rysy patří široký a plochý obličej a výrazné čelo.
- Koolen-de Vriesův syndrom (mírná až středně těžká mentální retardace, svalová slabost, záchvaty, přátelskost). Charakteristické rysy: dlouhý obličej s vysokým čelem, odstávající uši, šikmé oči, vysoká pohyblivost kloubů, vrozené srdeční vady.
- Phelanův-McDermidův syndrom (příznaky: mentální retardace, poruchy řeči nebo nedostatek řeči). Charakteristické znaky: velké ruce s vyvinutými svaly, svalová slabost od narození, slabé pocení.
Patologie, jako je deficit adenylsukcinátu, syndrom autozomálně recesivní mentální retardace 1, syndrom duplikace chromozomu 2q23.1, syndromy haploinsuficience genů FOXG1, STXBP1 nebo MEF2C a některé další, se mohou „chlubit“ příznaky podobnými Angelmanovu syndromu.
Úkolem lékaře je stanovit přesnou diagnózu, odlišit Angelmanův syndrom od patologií s podobnými příznaky a předepsat účinnou léčbu, která je relevantní pro diagnostikované stádium onemocnění.
Léčba Angelmanův syndrom
Angelmanův syndrom je jednou z patologií, pro které medicína stále hledá účinnou léčbu. Etiologická léčba onemocnění je ve fázi vývoje různých metod a prostředků, z nichž mnohé dosud nebyly testovány na lidech. To znamená, že se lékaři prozatím musí omezit na symptomatickou terapii, která pomáhá nějakým způsobem zmírnit nezáviděníhodnou situaci dětí i dospělých s marionetovým syndromem, trpících epileptickými záchvaty, sliněním, hypotenzí a poruchami spánku.
Je tedy možné snížit frekvenci a sílu epileptických záchvatů pomocí správně zvoleného antikonvulzivního léku. Celý problém však spočívá v tom, že záchvaty u pacientů se SA se od běžných epileptických záchvatů liší tím, že se vyznačují několika typy záchvatů, což znamená, že stav lze zmírnit podáním několika léků najednou.
Nejoblíbenější antikonvulziva používaná k léčbě Angelmanova syndromu jsou: kyselina valproová, topiramát, lamotrigin, levetiracetam, klonazepam a léky na jejich bázi. Méně často se používají léky na bázi karmazepinu, fenytoinu, fenobarbitalu a ethosuximidu, protože některé z nich mohou vyvolat paradoxní účinek spočívající v zesílení a zvýšení frekvence epileptických záchvatů. K tomu dochází, pokud se lék používá jako součást monoterapie.
K léčbě slinění se obvykle používají dvě metody: medikamentózní (léky potlačující tvorbu slin) a chirurgická, která zahrnuje reimplantaci slinných kanálků. V případě sankce se však tyto metody považují za neúčinné a otázka zůstává otevřená. Rodiče a ti, kteří se o takové pacienty starají, musí této problematice věnovat zvláštní pozornost, protože pacienti sami obvykle slinění nekontrolují a někteří se o sebe prostě nedokážou postarat.
Dalším problémem je krátká délka spánku. Děti s Angelmanovým syndromem často spí maximálně 5 hodin, což má negativní dopad na fungování celého těla. Snadno vzrušivé, aktivní děti, které milují hry a komunikaci (i když se snaží omezit na neverbální metody), jsou během dne znatelně unavené. Aby si tělo dobře odpočinulo, potřebuje hluboký a plný spánek, ale právě v tom je háček.
Zdálo by se, že sedativní léky (fenothiaziny a atypická antipsychotika), které zklidňují nervový systém, by měly být dostatečné ke zlepšení spánku u pacientů s podrážděností. V případě AS je však užívání těchto léků plné negativních účinků. Lékaři proto stále preferují mírné léky na spaní, jako je melatonin (přírodní hormonální lék založený na hormonu spánku), který se pacientům podává hodinu před spaním v množství 1 tablety, a difenhydramin. Frekvenci podávání a dávkování určuje lékař v závislosti na stavu a věku pacienta.
Pacienti s Angelmanovým syndromem mívají někdy problémy s trávením a stolicí. Stolici můžete zlepšit projímadly (nejlépe bylinnými).
Nebo můžete k problému přistupovat jinak, jak to udělali američtí lékaři, na základě některých metod léčby autismu, protože mnoho symptomů charakteristických pro AS je charakteristických i pro autismus (impulzivita, mimovolní pohyby, opakující se akce, deficit pozornosti, komunikační problémy atd.). Bylo zjištěno, že podání hormonu sekretinu, který normalizuje trávení a stolici, má pozitivní vliv na pozornost pacientů, a oxytocin pomáhá zlepšit kognitivní schopnosti a paměť dítěte a korigovat chování.
Pravda, samotné hormony nestačí, zejména pokud jde o děti. U Angelmanova syndromu je indikována behaviorální terapie, práce s psychologem a logopedem (výuka neverbálních komunikačních metod a znakového jazyka). Vzdělávání takových dětí by mělo být založeno na individuálním programu za účasti speciálně vyškolených učitelů, psychologa a rodičů. Bohužel to není možné všude a rodiny zůstávají se svým problémem samy.
Vzhledem k tomu, že mnoho mladých pacientů s AS trpí nízkým svalovým tonem a problémy s klouby, je velká pozornost věnována fyzioterapii. Nejčastěji se lékaři uchylují k parafínovým aplikacím, elektroforéze a magnetoterapii.
Aktivní tonizující masáž a speciální cvičení léčebné tělesné výchovy pomohou nemocnému dítěti po chvíli postavit se na nohy a sebejistě chodit. V tomto ohledu je obzvláště užitečná aquagymnastika, která se doporučuje při SA v chladné vodě. Zvyšuje svalový tonus a učí dítě ovládat své tělo a koordinovat pohyby.
Antikonvulzivní léčba
Nejnebezpečnějším příznakem Angelmanova syndromu jsou záchvaty podobné epilepsii. Tento příznak se pozoruje u 80 % pacientů, což znamená, že všem je třeba předepsat účinnou antikonvulzivní léčbu.
Léčba epileptických záchvatů se provádí pomocí vitamínů a antikonvulziv. U Angelmanova syndromu, doprovázeného křečovým syndromem, budou užitečné vitamíny skupiny B, stejně jako vitamíny C, D a E. Samostatné předepisování vitamínové terapie je však v tomto případě velmi nebezpečné, protože nekontrolovaný příjem vitamínů může snížit účinnost antiepileptik a vyvolat nové, závažnější a prodloužené záchvaty.
Výběr antikonvulziv a předepsání jejich účinného dávkování by měl provádět také odborný lékař. Ten také rozhodne, zda bude stačit jeden lék, nebo zda bude pacient muset užívat 2 či více léků dlouhodobě.
Většině pacientů lékaři předepisují léky s kyselinou valproovou (kyselina valproová, Depakine, Convulex, Valparin atd.), které zabraňují záchvatům a zlepšují náladu a duševní stav pacientů.
Kyselina valproová je dostupná ve formě tablet, sirupu a injekčních roztoků. Nejoblíbenějším lékem je lék s prodlouženým uvolňováním "Depakine" v tabletách a jako roztok pro intravenózní podání. Dávkování léku určuje lékař individuálně v závislosti na hmotnosti, věku a stavu pacienta.
Lék se užívá během jídla 2 až 3krát denně. Průměrná denní dávka je 20-30 mg na 1 kilogram hmotnosti pacienta, maximální je 50 mg/kg denně.
Kontraindikace k použití. Nepoužívejte v případě dysfunkce jater a slinivky břišní, hemoragické diatézy, hepatitidy, porfyrie a přecitlivělosti na léčivo.
Mezi nežádoucí účinky patří třes rukou, poruchy trávení a stolice a změny tělesné hmotnosti.
"Topiramát" je také lékem volby pro SA. Vyrábí se ve formě tablet a používá se jak v monoterapii, tak v kombinaci s jinými léky.
Způsob podání a dávkování. Tablety se užívají perorálně bez ohledu na příjem jídla. Počáteční denní dávka pro dospělé je 25–50 mg, pro děti 0,5–1 mg/kg. Každý týden se dávka zvyšuje dle pokynů lékaře.
Lék by se neměl užívat během těhotenství a kojení, stejně jako v případě přecitlivělosti na jeho složky. Lék má mnoho různých vedlejších účinků.
Léky, které může lékař předepsat na Angelmanův syndrom: Clomazepam, Rivotril, Lamotrigin, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra atd.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Tradiční medicína a homeopatie
Tradiční medicína, stejně jako homeopatické přípravky, je samozřejmě relativně bezpečná, ale účinnost takové léčby Angelmanova syndromu lze považovat za kontroverzní.
I když lidová léčba může v některých věcech stále pomoci. Mluvíme o zastavení epileptických záchvatů. V tomto ohledu může být bylinná léčba docela účinná.
Dobrý účinek poskytuje léčivý sběr na bázi pivoňky, lékořice a okřehku (složky se užívají ve stejném množství). Byliny je třeba rozemlít na mouku. Po 2 týdnech od zahájení užívání si můžete všimnout výrazného snížení frekvence záchvatů.
Levandulový odvar (1 čajová lžička na sklenici vroucí vody) je také užitečný při křečích. Směs se vaří 5 minut a louhuje se půl hodiny. Lék se užívá na noc po dobu 14 dnů.
Vodný (nebo alkoholový) nálev z mateří dřeně je považován za účinný při epileptických záchvatech.
Z homeopatických přípravků k prevenci záchvatů u Angelmanova syndromu můžete použít léky na bázi heřmánku a motherworm, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Je však třeba vzít v úvahu, že účinné a bezpečné dávky léků v každém konkrétním případě může předepsat pouze homeopatický lékař.
Prevence
Jak čtenář pravděpodobně již pochopil, medicína zatím nedokáže zabránit genovým mutacím a dalším chromozomálním abnormalitám a situaci ani napravit. To se může stát komukoli, protože děti s Angelmanovým syndromem se rodí zdravým rodičům a genetika, která je v současnosti jedním z nejméně prozkoumaných oborů medicíny, to zatím nedokáže vysvětlit.
Jediné, co se dá udělat, je zodpovědně přistupovat k plánování těhotenství, včas se zaregistrovat a podstoupit vyšetření. Ale opět, takové opatření bude spíše výchovné než preventivní, jako jakékoli vyšetření. Mladí rodiče ale budou předem vědět, na co se mají připravit, a v případě kladné odpovědi se rozhodnou, zda si mohou vzít na sebe takovou zodpovědnost, jako je výchova nemocného dítěte.
Předpověď
Prognóza Angelmanova syndromu závisí na povaze chromozomální abnormality a včasnosti její detekce. Nejvíce jsou postiženy ty děti, jejichž chromozom 15 obsahuje „mezery“ v genech (delece). Pravděpodobnost, že tito pacienti budou chodit a mluvit, je extrémně nízká. Jiné případy lze napravit pečlivým přístupem a láskou k vašemu dítěti.
Takoví pacienti se bohužel nebudou moci stát plnohodnotnými členy společnosti, a to i přesto, že zdaleka nejsou hloupí, rozumí řeči a jejímu významu. S komunikací však budou mít problémy po zbytek života. Pacienty lze učit znakovou řeč od dětství, ale nelze je nutit ke komunikaci pomocí slov. Slovní zásoba „mluvících“ pacientů je omezena na minimum slov používaných v běžném životě (5–15 slov).
Pokud jde o délku života a celkový zdravotní stav pacientů s Angelmanovým syndromem, zde se čísla pohybují kolem průměrných hodnot. V dospělosti se pacienti většinou potýkají se zdravotními problémy, jako je skolióza a obezita, které při správném přístupu k léčbě neohrožují život.