
Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
Keratodermie: příčiny, příznaky, diagnóza, léčba
Lékařský expert článku

Naposledy posuzováno: 07.07.2025
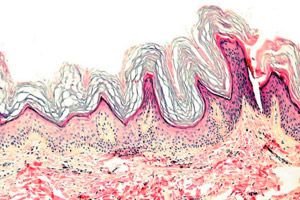
Keratoderma je skupina dermatóz charakterizovaných narušením procesu keratinizace - nadměrnou tvorbou rohovin zejména na dlaních a chodidlech.
Příčiny a patogeneze onemocnění nebyly dosud zcela objasněny. Výzkum prokázal, že keratodermie jsou způsobeny mutacemi v genech kódujících keratin 6, 9, 16. Velký význam v patogenezi hraje nedostatek vitaminu A, hormonální dysfunkce, zejména pohlavních žláz, bakteriální a virové infekce. Jsou jedním z příznaků dědičných onemocnění a nádorů vnitřních orgánů (parapsoriatické keratodermie).
Příznaky. Rozlišuje se difúzní (Unna-Tostova keratodermie, Meledovská keratodermie, Papillon-Lefevreova keratodermie, mutilující keratodermie a syndromy, které zahrnují difúzní keratodermii jako jeden z hlavních příznaků) a fokální (diseminovaná skvrnitá keratodermie Fischer-Buschkeho, akrokeratoelastoidóza Kostiho, omezená keratodermie Bruhauer-Franzesthestiho, lineární keratodermie Fuchse atd.) keratodermie.
Winy-Tostova keratodermie (synonyma: vrozená ichtyóza dlaní a chodidel, Winy-Tostův syndrom) se přenáší autozomálně dominantním způsobem. Dochází k difúzní nadměrné keratinizaci kůže dlaní a chodidel (někdy pouze chodidel), která se vyvíjí v prvních dvou letech života. Kožní patologický proces začíná mírným ztluštěním kůže dlaní a chodidel ve formě pruhu erytému lividní barvy na hranici se zdravou kůží. Postupem času se na jejich povrchu objevují hladké, nažloutlé rohovité vrstvy. Léze se zřídka šíří na hřbet zápěstí nebo prstů. U některých pacientů se mohou tvořit povrchové nebo hluboké praskliny a je zaznamenán lokální hyperhidróza. U pacienta pozorovaného autorkou trpěli Winy-Tostovou keratodermií strýc z matčiny strany, bratr a syn.
Jsou popsány případy poškození nehtů (ztluštění), zubů a vlasů u keratodermy Winy-Tost v kombinaci s různými kosterními anomáliemi a patologií vnitřních orgánů, nervového a endokrinního systému.
Histopatologie. Histologické vyšetření odhaluje výraznou hyperkeratózu, granulózu, akantózu a malé zánětlivé infiltráty v horní vrstvách dermis. Diferenciální diagnostika. Onemocnění je nutné odlišit od jiných typů keratodermie.
Meledova keratodermie (synonyma: Meledova choroba, vrozený progresivní akrokeratom, Siemensova palmoplantární transgradientní keratóza, Kogoyova hereditární palmoplantární progresivní keratóza) se dědí autozomálně recesivně. Tato forma keratodermie se vyznačuje silnými, žlutohnědými rohovitými vrstvami s hlubokými prasklinami. Podél okrajů léze je viditelný fialově fialový okraj široký několik milimetrů. Proces se obvykle šíří na hřbet rukou a nohou, předloktí a holeně. Většina pacientů pociťuje lokální hyperhidrózu. V tomto ohledu se povrch dlaní a chodidel stává mírně vlhkým a pokrytým černými tečkami (vývody potních žláz).
Onemocnění se může rozvinout ve věku 15-20 let. Nehty ztlušťují a deformují se.
Histopatologie. Histologické vyšetření odhalí hyperkeratózu, někdy akantózu a chronický zánětlivý infiltrát v papilární dermis.
Diferenciální diagnóza. Melelovou keratodermii je nutné odlišit od Unna-Tostovy keratodermie.
Keratoderma Papillon-Lefevre (synonymum: palmoplantární hyperkeratóza s parodontitidou) se dědí autozomálně recesivně.
Onemocnění se projevuje ve 2.–3. roce života. Klinický obraz onemocnění je podobný Melelově chorobě. Charakteristické jsou navíc změny na zubech (abnormality v prořezávání mléčných a stálých zubů s rozvojem kazu, gingivitida, rychle postupující parodontóza s předčasnou ztrátou zubů).
Histopatologie. Histologické vyšetření odhaluje ztluštění všech vrstev epidermis, zejména rohovité vrstvy, a nevýznamné buněčné shluky lymfocytů a histiocytů v dermis.
Diferenciální diagnóza. Toto onemocnění je třeba odlišit od jiných keratodermií. Důležitým rozlišovacím znakem je charakteristická zubní patologie, která se u jiných forem hereditárních difuzních keratodermií nevyskytuje.
Keratoderma mutilans (synonyma: Fonwinkelův syndrom, hereditární mutilující keratom) je typ difúzní keratodermy dědičné autozomálně dominantním způsobem. Vyvíjí se ve 2. roce života a je charakterizována difúzními rohovitými ložisky na kůži dlaní a chodidel s hyperhidrózou. Postupem času se na prstech tvoří provazcovité rýhy, které vedou ke kontrakturám a spontánní amputaci prstů. Folikulární keratóza se projevuje na hřbetech rukou, stejně jako v oblasti loketních a kolenních kloubů. Nehtové ploténky jsou změněny (často jako hodinková sklíčka). Byly popsány případy hypogonadismu, rubínové alopecie, ztráty sluchu, pachyonychie.
Histopatologie. Histologické vyšetření odhaluje těžkou hyperkeratózu, granulózu, akantózu a malé zánětlivé infiltráty v dermis, sestávající z lymfocytů a histiocytů.
Diferenciální diagnostika. Při diferenciaci mutilující keratodermy od jiných forem difuzní keratodermy je třeba v první řadě vzít v úvahu mutilační efekt, který není pro jiné formy typický. Při provádění diferenciální diagnostiky všech forem difuzní keratodermy je nutné mít na paměti, že může být jedním z hlavních příznaků řady dědičných syndromů.
Léčba. Neotigazon je indikován v celkové terapii keratodermie. Dávka léku závisí na závažnosti procesu a je 0,3-1 mg/kg hmotnosti pacienta. Při absenci neotigazonu se doporučuje vitamín A v dávce 100 až 300 000 mg denně po dlouhou dobu. Zevní terapie spočívá v použití mastí s aromatickými retinoidy, keratolytických a steroidních látek.
 [
[ Co tě trápí?
Co je třeba zkoumat?
Jak zkoušet?